

he drug discovery and development field established over the years a range of in vitro and in vivo tools to model host-drug interactions and better estimate the pharmacodynamics (PD), pharmacokinetics (PK), and toxicity of drug candidates in humans. However, the microbiome’s role was generally ignored (see article in September Global Forum). The range of effects that the gut microbiome might have on administered drugs can produce unexpected outcomes on PK, PD, and toxicity. In turn, drugs may impact the gut microbiome and its interactions with the host cells (intestinal epithelial cells, immune cells, nerve cells, and enteroendocrine cells), perturbing normal body homeostasis. At the extreme end, such as with antibiotics, disruption to the microbiome can lead to dysbiosis and aggravate diseases.
Studying the Drug-Microbiome Interactions in Vitro
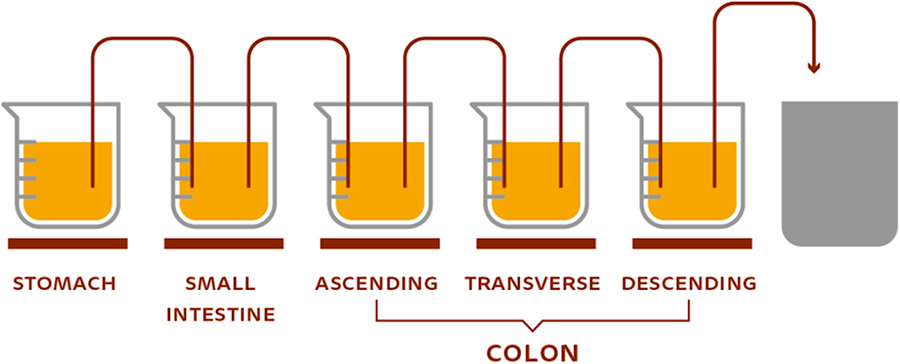
In summary, despite the absence of a physiological host environment, multicompartment in vitro systems can maintain a humanlike gut microbiota and specific activities observed in humans, and allow the study of drug-microbiome interactions without interference from the host. The system is even more predictive when the mucosal environment, in addition to the luminal environment, is incorporated by adding mucin to the model. In vitro models offer additional advantages as they are standardized and generate results with high reproducibility. Also, there are no ethical constraints. Finally, despite its important role in understanding drug-microbiome interactions, it is crucial to combine this technology, very useful for screening purposes, with lower-throughput approaches that more closely resemble the in vivo situation, as interactions between drugs and microbes identified in vitro need to be validated in the host context.
Studying the Drug-Microbiome-Host Triad in Vitro and in Vivo
An important advancement in the study of drug-microbiome-host triads was the development of organoids by differentiating stem cells into specific intestinal cell types, such as enterocytes, goblet cells, and Paneth cells, that formed spherical crypt-like structures. When combined with microfluidic technology, these cells form a gut-on-a-chip device that models the whole intestine. This type of in vitro technology holds a promising future by transforming our mechanistic understanding of the drug–microbiome–host triad.
In pre-clinical development, the ultimate goal is to understand the drug-microbiome-triad in a whole organism, generally in rodents, the standard model used in drug and microbiome research for many years. For example, in cancer immunotherapy, the gut microbiota plays an important role in host response to treatment. And research with mice has shown that the effect of PD1 and PDL1 checkpoint inhibitors against melanoma can be enhanced with increased abundance of Bifidobacterium species.
When it comes to drug metabolism, there are few strategies to separate host and microbiome contributions, such as the comparison of drug and drug metabolites’ systemic exposures between conventional and germ-free animals, conventional, germ-free, and humanized animals (colonized with human gut microbiota), and through combinatorial therapies, i.e., the drug under investigation with and without antibiotics. All of these efforts aim at teasing out the influence of the microbiome on the drug and drug metabolites’ PK, PD, and toxicity.
Call to Action
It is imperative that the drug discovery and development field incorporates systematic approaches, such as the ones briefly described herein, to understand drug-microbiome interactions and the drug-microbiome-host triad, complementing the current in vitro and in vivo arsenal routinely employed to model host-drug interactions. This would (i) improve compound selection in preclinical development, (ii) better define specific patient populations, taking microbiome composition and activity into account, (iii) reduce or at least better understand interpersonal variability, and (iv) bridge the gap between pre-clinical and clinical results, ultimately leading to safer and more efficacious therapies. As added benefits, it would pave the way to repurposing nonantibiotic drugs with antimicrobial activity to develop nonresistant antibiotics and present a new perspective on the unknown origins of efficacy and side effects of many drugs in the market.