

Sharing the Regulatory Workload
he ACSS Consortium is an international cooperation between medium-sized regulatory authorities that include the Therapeutic Goods Administration of Australia (TGA), Health Canada (HC), Singapore’s Health Sciences Authority (HSA), and the Swiss Agency for Therapeutic Products (Swissmedic).
The work sharing consortium was formed in 2007 to reduce duplication of efforts and ensure timely consumer access to safe and effective therapeutic products, addressing the increased workload, greater complexity of issues, and limited resources agencies face these days. Currently, the ACSS Consortium has a number of active working groups in place, focusing on new chemical entities, complementary health products, IT architecture, and generic medicines, to name just a few. Each working group is responsible for its own work plan under the governance of the Heads of Agencies.
The Consortium’s Generic Medicines Working Group (GMWG) has successfully completed its assessment of the first application submitted through a new work sharing model, the Generic Medicines Work Sharing Trial (GMWST).
The Generic Medicines Work Sharing Trial
In applications for generic medicines, the GMWG has historically focused its efforts on information sharing and regulatory convergence in the areas of quality and bioequivalence. The GMWG has now moved on to exploring work sharing opportunities among Consortium regulatory agencies. As a result of these efforts, the ASCC Consortium launched a new work sharing model similar to the European Decentralised Procedure (DCP), the Generic Medicines Work Sharing Trial (GMWST), to coordinate the assessment of a generic medicine application that has been submitted to multiple agencies.
In this Trial, a single dossier, including the common Modules 2-5 and the country-specific Module 1, is simultaneously submitted to all participating agencies. Similar to the European DCP model, one agency is appointed as a Reference Regulatory Agency (RRA) whereas the other agencies are considered Concerned Regulatory Agencies (CRAs). The RRA undertakes an initial assessment of Modules 2-5, while the CRAs simultaneously undertake Module 1 assessments.
Upon completion of the initial RRA assessment, all CRAs review the RRA’s assessment reports (ARs) and its proposed list of questions to the Applicant (LoQ) and have the opportunity to include additional questions. The consolidated LoQ is then sent to each local affiliate of the Applicant for response. The RRA subsequently assesses the applicant’s response and produces a second AR for the CRAs to review. Each participating agency must then make its final regulatory decision within the framework of its respective national procedures.
To guide this work sharing trial, a series of documents was developed, including Operational Procedures, Questions and Answers, and an Expression of Interest (EOI) form. Figure 1 illustrates the timeline for different milestones of the trial.
First Application
The purpose of the first application within the trial was to work through the practicalities of undertaking a single assessment that will support regulatory decision-making within each jurisdiction in accordance with the respective regulatory requirements.
Teva Pharmaceuticals, a global generic pharmaceutical company with experience in filing applications with all the ACSS agencies, participated in the first trial. The application was submitted to the participating regulatory agencies of Australia, Canada, and Switzerland, while Singapore’s HSA served as an observer. The application concerned an oral, immediate-release tablet, containing a non-compendial, semi-synthetic, and poorly soluble drug substance. The application was supported by two bioequivalence (BE) studies: a fed BE study against the EU reference product (RP) and a three-arm fasting BE study against the Canadian and EU RPs. These studies, together with a justification for using a foreign RP rather than the domestic RP, were required to satisfy all three agencies’ clinical and BE requirements.
The trial started with the submission of a completed EOI form stating the company’s intent to participate in the GMWST, and a confirmation that the application complied with all of the listed eligibility criteria. In addition, the company detailed any differences between the data in Modules 2-5 submitted to each participating agency in a table entitled “Summary of Quality and Biopharmaceutic Studies Differences.” In this case, the differences included additional documentation for HC (master and executed batch records); results from the comparative analysis of the European, Swiss, and Australian RP formulations; differences in submitted strengths (one strength was omitted in one country); and the different sources of the RPs used for the BE studies as described above.
The Applicant and ACSS Consortium held a pre-submission teleconference two months prior to the filing of the application to discuss and confirm the logistics and expectations related to requirements, timelines, and process; to allow the agencies to respond to any queries from the Applicant; and to ensure that the dossier would be complete and acceptable for assessment prior to submission in each jurisdiction. The dossier was subsequently submitted to the TGA, HC, and SMC in the first half of 2016. For this first application, the TGA was designated as the RRA, while HC and SMC were the CRAs. Following the assessment, the LoQ was simultaneously sent to each local affiliate 55 days after the application was accepted. Two additional rounds of questions and responses resolved all remaining issues.
Comparative Timelines
As stipulated in the GMWST Operational Procedures, the agencies followed their own national procedures once the assessments were complete. Final approval of the application therefore occurred on different dates in each jurisdiction.
- SMC approved the application in March 2017, 260 days after acceptance for assessment and considerably shorter than the agency’s standard target of 480 days for a generic drug product.
- TGA approved the application in April 2017, 270 days after acceptance for assessment, close to its standard target time of 274 days for a generic drug product.
- Approval by HC occurred in April 2017, 289 days after acceptance for assessment and shorter than the 375-day target for a generic drug product.
Lessons Learned
- Due to the aggressive trial timelines and deviations from standard national procedures, good communication and coordination within the ACSS agencies, and between the agencies and the Applicant, were crucial for this trial’s success.
- The GMWST Operational Procedures did not include details for managing multiple rounds of questions but will be updated to accommodate this scenario. In addition, participating agencies learned that the RRA must include sufficient detail in the ARs to enable CRAs to perform their appropriate peer assessment.
- Industry feedback indicated that the initial scope of potential applications for the trial (e.g., immediate-release solid orals and solutions) was limited.
- Finally, while the application was more complex than initially anticipated, it provided a valuable learning opportunity.
Conclusions
A major benefit was that the participating agencies became more confident as they gained insights into how the other agencies assess each aspect of the application.
The GMWG has decided to continue with the work-sharing trial and widen the scope of potential products to include generic product applications for any dosage form.
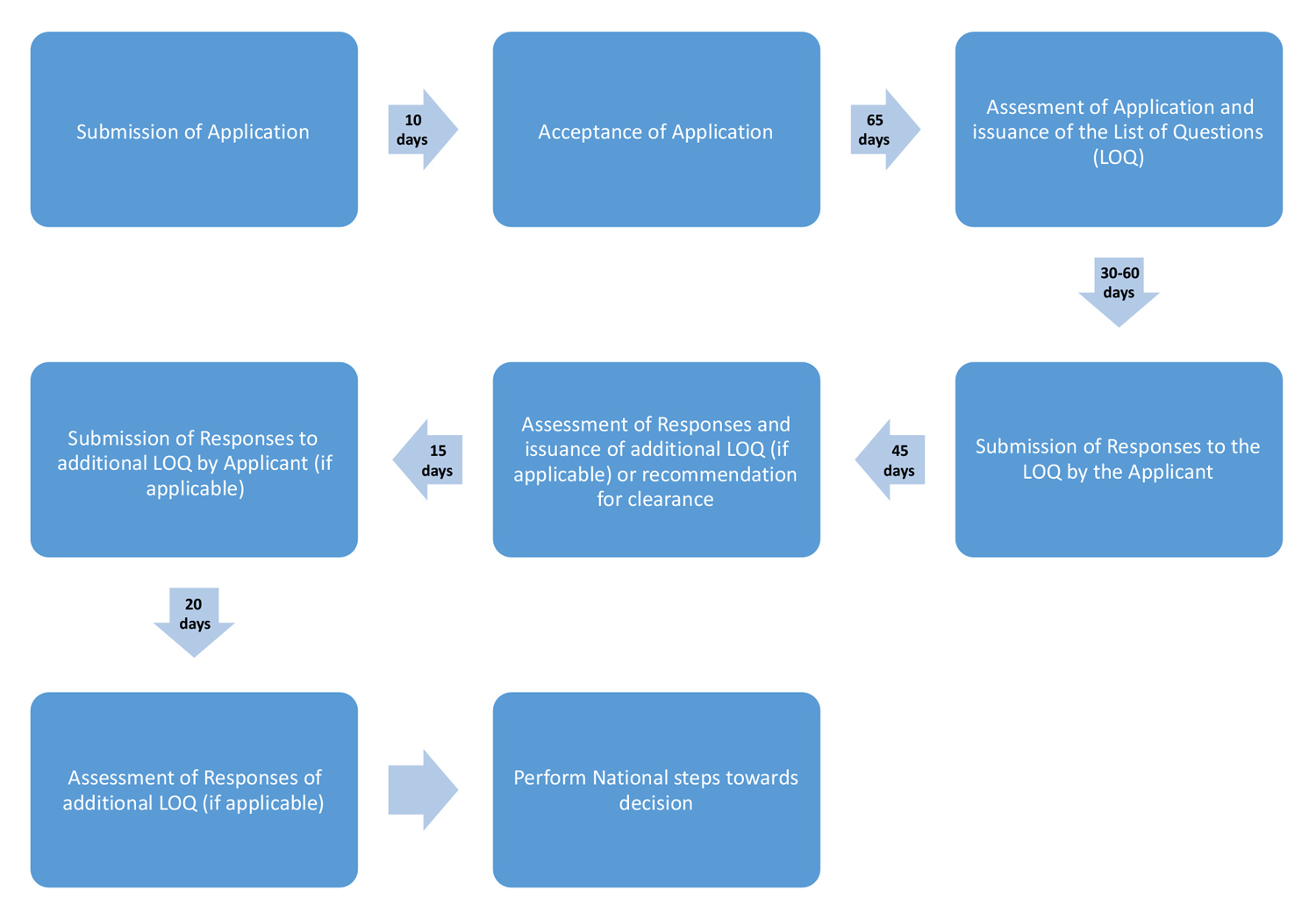
Figure 1 – Schematic representation of expected milestones and timelines
Health Canada and Swissmedic based their reviews on the TGA evaluation reports and did not conduct an in-depth review of the application documentation. As a result, they were able to dedicate more time to key quality and clinical assessment, indicating that a CRA workloads may be significantly reduced in future applications.
Successful completion of the first GMWST application demonstrates that this work-sharing model has the potential to benefit both industry and regulators. This trial strengthened collaboration among regulatory authorities, fostered greater understanding of technical requirements, and promoted regulatory convergence. Industry will likely benefit from a reduction in regulatory burden (e.g., through filing common dossiers), more timely regulatory decisions, concurrent marketing authorisations to multiple markets, and an opportunity to contribute to advancing regulatory innovation. This truly unique, global collaboration will continue with an expanded scope, providing a model that can be adopted on a larger scale.
Please direct queries relating to the GMWST to networking@swissmedic.ch.
About the Authors

Left to right: Craig Simon, Health Canada, Canada; Christopher Crane, Therapeutic Goods Administration, Australia; Arno Nolting, Swissmedic, Switzerland; Chantal Pfäffli, Swissmedic; Richard Weissmahr, Swissmedic; Claire Rodriguez, Health Sciences Authority, Singapore; Michael Harding, Therapeutic Goods Administration, Australia; Subin Sankarankutty, Health Sciences Authority, Singapore; and Gary Condran, Health Canada. Not pictured: Ricarda Meincke, Swissmedic.
Lead author Chantal Pfäffli received her MS in Pharmaceutical Science from the University of Basel. After serving as a pharmacist for three years, she started working with Swissmedic where she is responsible for oncological products and coordinates international activities of ACSS and IPRP (generics).