

Adaptive Biotechnologies
he T cells circulating in an individual’s blood represent a record of their prior immune exposures. Reading that record is an elusive goal but an important one: knowing prior immune exposures can predict future risk of severe disease or inform treatment decisions. Through our immunosequencing platform, we have amassed a database of T-cell repertoires from tens of thousands of individuals. Along with our partners at Microsoft, we’ve harnessed that enormous database to learn a great deal about a person’s exposure history from a sample of their blood.
T-Cell Repertoires are Ledgers of Immune Exposures
You get COVID. It’s your first exposure to SARS-CoV-2. As you become symptomatic, your T cells (a critical component of your adaptive immune response, each expressing a unique T-cell receptor, or “TCR”), pass through your lymph nodes. Among the millions of T cells in your blood, hundreds or thousands recognize SARS-CoV-2. Over the next week or two, these T cells activate and help eliminate the virus from your body.
As they respond to the virus, those T cells clonally “expand” (multiply) and make up a much larger share of your total T-cell repertoire than before. Afterward, many of those expanded clones will remain. Next time you encounter SARS-CoV-2, they’ll deliver a quicker, more efficient response.
Your immune system contains a vast number of unique T cell clones (perhaps around 108). When we extract and sequence T-cell DNA from a blood sample, we’re only capturing the TCRs from some of those T cells, routinely around 500,000. If we sequenced your T-cell repertoire before and after your bout with COVID, we’d sequence many more of those SARS-CoV-2-activated T cells in your post-COVID sample.
If you infect a friend with SARS-CoV-2, by the time they recover, their T-cell repertoire will also contain many SARS-CoV-2-recognizing TCRs. Some of theirs will likely be identical to some of yours. When we look at a large number of T-cell repertoires, we tend to see some of the same TCRs in many different people; we call those “public” TCRs. Many TCRs are public because they represent a shared response to an antigen from a prevalent exposure like SARS-CoV-2. That means they tend to occur in people who have been exposed to the antigen they respond to, and not in people who haven’t. And that’s true of many different immune exposures, all at once.
Figure 1 illustrates this idea. All three panels contain the same data, a matrix of 100 public TCRs that are present (black) or absent (white/color) in 100 different people’s repertoires. In the first panel, the TCRs and the people are in random order. If we rearrange the TCRs by “occurrence similarity,” so that each TCR is closest to the other TCRs whose pattern of donor occurrence is the most similar, a pattern becomes clear in the second panel: there are four distinct groups of TCRs, representing responses to four different exposures. In the third panel, we cluster the people (rows) by how many exposure-responding TCRs they share: i.e., by which exposures they have had.

And there’s one more major complication: each TCR responds to a peptide “presented” by a human leukocyte antigen (HLA) protein. You can think of the HLA as a hand that’s holding out the peptide for TCRs to interact with. Each HLA is capable of presenting some peptides but not others. Each person expresses just one of the hundreds of importantly different versions (“alleles”) of the eight HLA proteins, each capable of presenting different antigenic peptides. So, you and your friend both got COVID, but you likely only share SARS-CoV-2-responding TCRs presented by the HLA alleles you have in common. Unless you’re very closely related, that’s very few.
How can we tackle all this complexity to find groups of “public” (or shared) TCRs responding to the same exposure?
Finding T-Cell Signatures of Exposure
We start with T-cell repertoires from about 30,000 people. To build a matrix of TCRs in repertoires like the one in Figure 1, we first need to restrict ourselves to the context of antigens presented by a single HLA protein.
We’ve previously demonstrated that we can statistically determine which HLA protein (allele) is presenting the antigens to which many “public” TCRs respond and which individuals have which HLA alleles. Therefore, we can build a separate matrix for each HLA allele, considering only TCRs associated with the allele and individuals who have that allele. We computationally train a separate model for each HLA-specific matrix to find clusters of co-occurring TCRs. Each such “HLA-COcluster” nominally contains the TCRs responding to antigens from a single exposure, presented by a single HLA allele.
We next combine the TCR clusters representing the same immune exposure from all the different HLA contexts. To do that, we cluster the HLA-COclusters from all HLA contexts together, based on which people those TCRs occur in. Each of the resulting “ECOclusters” (Exposure Co-Occurrence clusters) putatively represents the public TCR response to a single exposure, comprising TCRs responding to antigens presented by many HLA alleles. Figure 2 shows how we build ECOclusters.

For seven exposures (CMV, SARS-CoV-2, EBV, HSV-1, HSV-2, parvovirus, and Toxoplasma gondii), we obtained repertoires from thousands of individuals with known serological status for those exposures. For each exposure, for each ECOcluster, we statistically tested whether individuals with the exposure had more TCRs that are members of the ECOcluster than individuals without the exposure.
For each of the seven exposures, we found a single ECOcluster far more strongly exposure-associated than any other (Figure 3). Furthermore, a person’s count of the TCRs within that ECOcluster defined a very strong diagnostic model for the exposure.
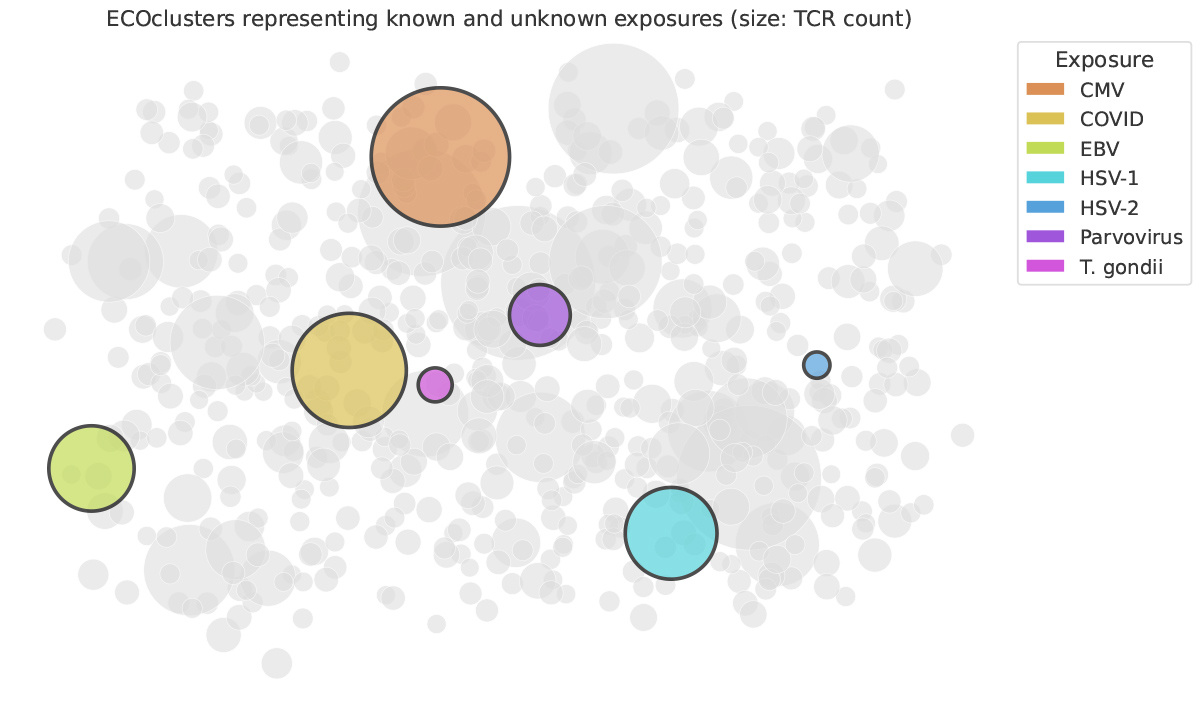
Now what?
We’re working on associating more ECOclusters with their exposures in two ways: by acquiring more repertoires from individuals of known exposure status, and by experimentally determining the antigens that ECOcluster TCRs bind to using an in vitro antigen-provoked T cell activation assay that we call MIRA. In the next few months, we anticipate finding the ECOclusters associated with about 30 more exposures. In the next few years, we expect to be able to diagnose exposure to dozens more pathogens from a single blood sample.
Furthermore, among individuals who have had a given exposure, ECOcluster response is a quantitative measure. In trials of vaccines, or of treatments like oncolytic viruses, we can use ECOclusters to quantify each patient’s response to treatment. We’re already using this capability to support Pharma studies. We also hope to use that quantitative measure to predict treatment outcomes, as well as to explore the relationships between prevalent exposures (like viruses) and serious illnesses (including autoimmune disorders such as Type 1 diabetes).
Each person’s T-cell repertoire is a ledger of their prior immune exposures. ECOclusters are a major step toward learning how to read that ledger.