

Director, Regulatory Policy & FDA Engagement
BioMarin Pharmaceutical, Inc.
uman gene therapy (GT) is a treatment approach that seeks to modify or manipulate a person’s genes to treat or cure disease. GT can work in several ways including by (1) replacing a disease-causing gene with a healthy copy of the gene; (2) inactivating a disease-causing gene that is not functioning properly; or (3) introducing a new or modified gene into the body to help treat a disease. GT products are being developed to treat cancer, hemophilia, and other genetic diseases.
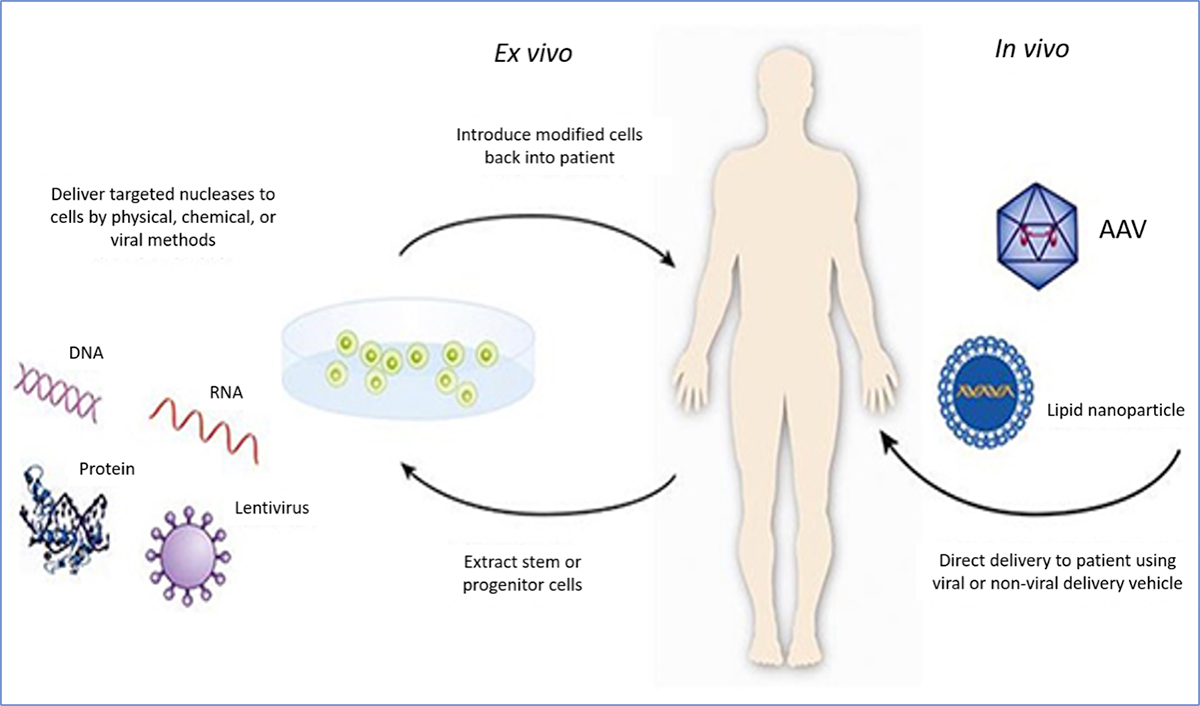
Figure 1. Types of gene therapy (from FDA Gene Therapy webpage)
GT holds the potential to provide transformative treatment options for diseases with high unmet medical need. In 2017, the FDA approved two Chimeric Antigen Receptor T cell (CAR-T) ex vivo GT products and one adeno-associated virus vector-based GT (Kymriah, Yescarta, and Luxturna). FDA has also received more than 500 investigational new drug applications (INDs) for GT products.
With advances in science and technology and the advent of GT, the regulatory environment must also evolve to keep pace with the science. Regulatory requirements for GT products should be clear and supportive of efficient development. Health authorities are developing the framework for development, evaluation, and regulation of gene therapy products.
To provide regulatory clarity in this rapidly evolving field, the FDA’s Center for Biologics Evaluation and Research (CBER) issued six guidances for industry on GT product development on July 11, 2018, adding to their existing portfolio of GT guidances. The draft guidances reflect experience gathered over the last decade with GT product development. The suite of six GT guidances includes three disease-specific draft guidances on (1) rare diseases, (2) hemophilia, and (3) retinal disorders, as well as three topic-specific draft guidances on (4) chemistry, manufacturing, and controls (CMC) information for GT investigational new drug applications (INDs), (5) long term follow-up (LTFU) after administration of GT products for the collection of data on delayed adverse events following administration of a GT product, and (6) testing for replication-competent retrovirus during product manufacture and patient follow-up. The disease specific guidances are structured to address different aspects of drug development, e.g., CMC, nonclinical, and clinical development. These documents generally reflect a pragmatic approach to development that would support innovation while ensuring safety and efficacy. Brief highlights of some recommendations included in each guidance follow.
Human Gene Therapy for Rare Diseases
CMC considerations discussed in the draft guidance highlight that some aspects of development programs for rare diseases may make it challenging to follow traditional manufacturing strategies. FDA recommends a well-controlled manufacturing process to assess product Critical Quality Attributes as early as possible in the development program.
Because of the challenges in enrolling patients with rare diseases, FDA recommends that sponsors consider designing their first-in-human study to be an adequate and well-controlled investigation that can provide evidence of effectiveness to support a marketing application. This recommendation is a positive change that helps to streamline development of these advanced therapies for patients with rare diseases. The recommendation also addresses the challenges for rare diseases with small patient populations, and also can help in expediting drug development. This guidance recommends that endpoint selection should consider the pathophysiology, natural history, and aspects that are meaningful to patients.
Safety considerations recommend a staggered administration approach for first-in-human trials to limit the number of subjects exposed to unanticipated safety risk. Duration of long-term follow-up depends on the results of pre-clinical studies with the product, knowledge of the disease process, and other scientific information. FDA encourages sponsors to collect patient experience data and submit them in the marketing application.
Human Gene Therapy for Hemophilia
For CMC considerations, FDA recommends establishing a potency assay to assess the biological activity of the final product, with relevant lot release specifications, prior to initiating clinical trials intended to provide substantial evidence of effectiveness for a marketing application.
Considerations for Factor VIII/Factor IX activity measurements assessed by different clinical laboratory assays include a discussion of “discrepancies” between one stage clotting (OC) and chromogenic (CS) assays to measure factor activity and recommendations for evaluation of the assay discrepancies early during clinical development.
The guidance provides recommendations regarding endpoints for developing GT products for hemophilia. For traditional approval, a non-inferiority clinical trial design with Annualized Bleed Rate as the primary endpoint using within-subject comparison design is recommended. Notably, FDA also provides the option for accelerated approval using factor activity as a surrogate endpoint so far as discrepancies in factor assay results from various assays are understood, and the factor activity level is within the range of the normal population. A specific recommended factor activity range is not provided.
As with the draft guidance for gene therapy for rare diseases, sponsors are encouraged to collect patient experience data and submit them in the marketing application.
Human Gene Therapy for Retinal Disorders
It provides several CMC considerations, including final product formulation to meet the expected dose and volume requirement, endotoxin limit for intraocular delivery, product testing and release, and evaluation of the compatibility of the GT product and delivery system.
For pre-clinical studies, FDA provides a number of recommendations including that animals with more “human-like” eyes (such as rabbits, pigs, dogs, or nonhuman primates) be used to generate proof-of-concept data. Inclusion of the larger animals facilitates relevant experience with the surgical procedures and delivery systems intended for clinical use.
According to the guidance, first-in-human GT trials should enroll patients with severities of visual impairment that offer a favorable benefit-risk profile. Planned sequential administration of the GT product in both eyes, development of a rationale for which eye should receive GT first, and a carefully planned time interval between administrations in each eye is recommended. FDA recommends that when single administration may not be sufficient, the feasibility of repeat administration in the same eye should be explored. FDA also encourages sponsors to develop and propose novel endpoints to measure clinically meaningful effects in patients with retinal disorders.
Chemistry, Manufacturing, and Control Information for Human Gene Therapy Investigational New Drug Applications
Long Term Follow-Up (LTFU) After Administration of Human Gene Therapy Products
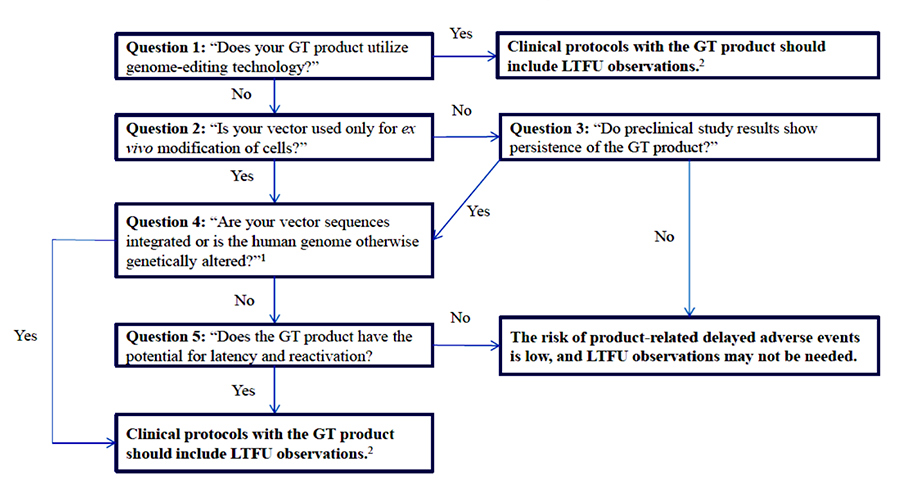
This draft guidance describes the Agency’s current recommendations for the conduct of LTFU studies, specifically the information and data required to support a sponsor’s rationale for the duration and design of a LTFU protocol when clinical trials are initiated. Notably, the guidance departs from FDA’s previous recommendation in their 2006 draft guidance for a blanket 15-year LTFU for all GT products. FDA now recommends a case-by-case approach of appropriate expectations for follow-up monitoring and for building in risk assessment as a key step. This risk-based approach reflects FDA’s increased confidence gained through experience with GT products in the last decade. FDA also notes that some rare GT products may not require LTFU observations. For adeno-associated virus (AAV) vectors, the FDA expects a follow-up of up to five years (two to five years). In other cases, such as those integrating vectors and genome editing products, FDA expects up to 15 years of follow-up. The guidance provides the following framework to assess the risk of GT-related delayed adverse events.
Testing of Retroviral Vector-Based Human Gene Therapy Products for Replication-Competent Retrovirus During Product Manufacture and Patient Follow-Up
This draft guidance provides recommendations regarding the testing for replication-competent retrovirus during the manufacture of retroviral vector-based gene therapy products, and during follow-up monitoring of patients who have received retroviral vector-based gene therapy products. Recommendations include the identification and amount of material to be tested as well as general testing methods. In addition, recommendations are provided for monitoring patients for evidence of retroviral infection after administration of retroviral vector-based gene therapy products. The draft guidance recommends a risk-based monitoring schedule that includes analysis of patient samples at the following time points: pre-treatment, followed by testing at three, six, and twelve months after treatment, and yearly for up to 15 years.