

Richmond Pharmacology
Alpharmaxim
omen may be represented in clinical drug trials, but they remain invisible in evidence. The gender data gap is real.
Why Women Remain Invisible in Evidence Generation
The gap persists due to unclear requirements, inconsistent enforcement, and a lack of early integration into protocol design or lifecycle evidence-generation planning. In an environment where teams prioritize timelines and costs for obvious reasons, the absence of female data and its eventual consequences can seem more abstract.
In one recent example, the consequences were clear: A widely prescribed insomnia drug required a post-marketing dose reduction when reports showed that women metabolized it more slowly, leading to next-morning impairment. When taking a standard dose, 15% of women still had an impairment level of the drug in their systems after 8 hours, compared to 3% of men.
Regulators mandated a labeling change only after these issues emerged. Would stronger early inclusion of women in clinical trials and clearer regulatory expectations have prevented this?
Where Evidence Breaks Down in Practice
Guidance exists, but evidence fails at three predictable points in the development and regulatory pathway for new medicines.
1. Inclusion without meaningful analysis
Women are increasingly included in clinical trials, but they are not always visible in decision-informing data, as shown in EMA dossier evaluations.
- Subgroup analyses are often underpowered
- Sex-specific findings are labeled as exploratory
- Protocols rarely pre-specify sex-based endpoints.
AI-supported signal detection can identify sex-specific safety patterns only if sex-disaggregated data is available. Otherwise, AI models risk reinforcing male-centric evidence and assumptions.
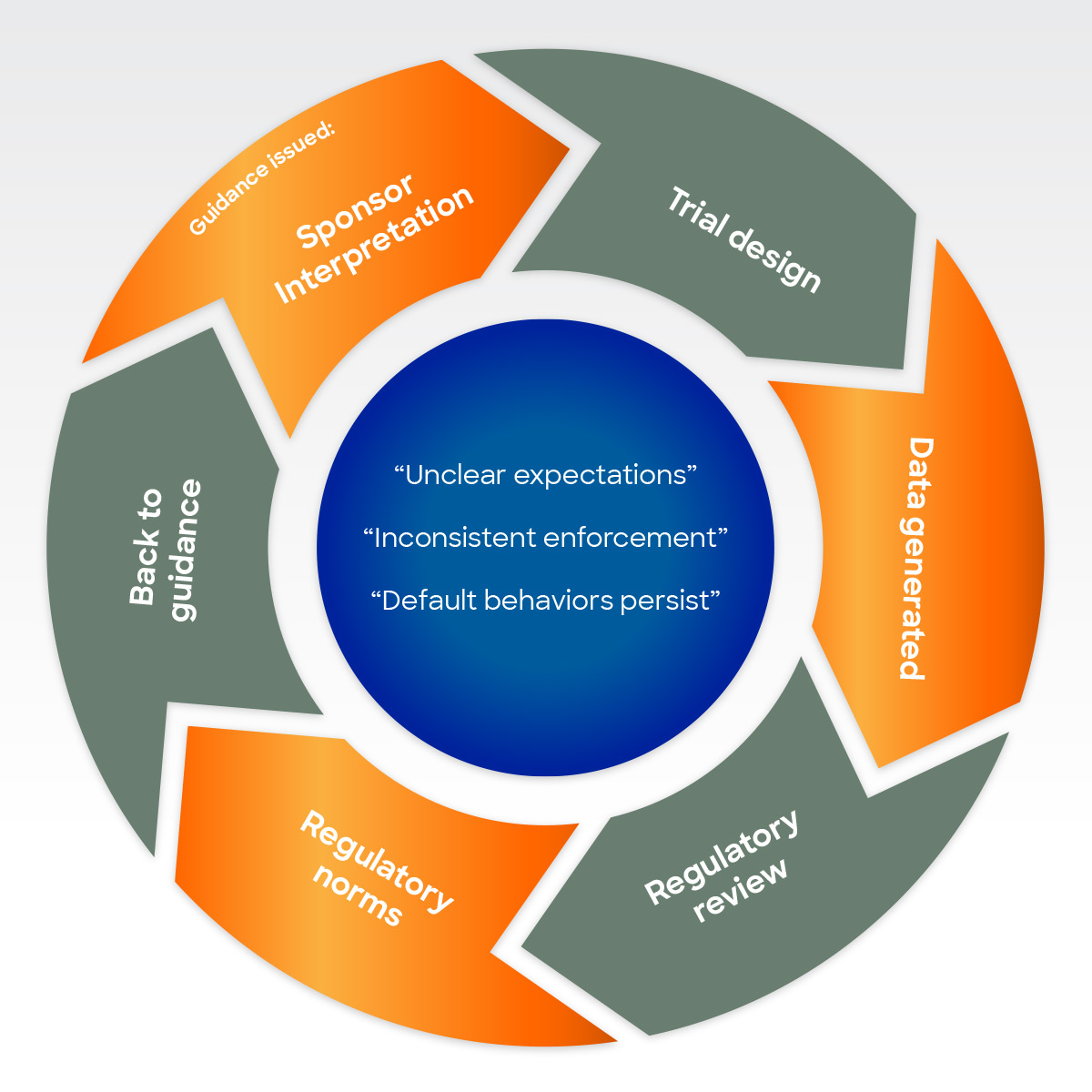
Behavioral defaults matter. When protocol templates treat sex-specific analysis as optional, omission becomes the norm. People rarely deviate from defaults, especially under time pressure. Reversing this requires changing the choice architecture:
- Make sex-specific endpoints the default in protocol design, requiring teams to actively opt out with justification that is reviewed alongside other core design decisions.
- Require justification when excluding them. Shift to “Why are we comfortable excluding them?” to raise the issue of the perceived cost of omission.
- Introduce structured checklists to guide analysis of pharmacokinetics, pharmacodynamics, and adherence differences.
These interventions increase intentionality, improve decision-making, reduce reliance on historical assumptions, and improve longevity and safety (through more accurate predictions of adverse events) for patients.
2. Early-phase evidence remains male-default
Phase 1 and phase 2 studies set foundations for dosing and safety, yet many programs still treat sex as a secondary variable rather than a core design consideration. This reflects the assumption that healthy male volunteers offer the simplest, lowest-risk study population, an assumption that persists partly due to status quo bias and availability heuristics.
While this may streamline operations, it introduces long-term risk:
- Dose selection may not reflect sex-based differences in metabolism.
- Adverse drug reactions may emerge disproportionately in women.
Post-marketing corrections become more likely. Shifting this tendency requires reframing risk. Sponsors must balance the perceived risk of including women in early-phase trials against the demonstrated risk of incomplete evidence.
Practical steps include:
- Designing early-phase cohorts with sex stratification
- Using scenario planning to anticipate downstream safety risks and model counterfactuals to inform early decisions
- Applying pre-mortem analyses to identify where sex differences could affect outcomes.
These approaches make future risks more visible and therefore more actionable, positioning sex-inclusive early planning as a rational response to cognitive bias rather than as a moral challenge.
3. Pregnant or lactating women remain excluded
Drugs are licensed without data for pregnant or lactating women, denying them informed choices. This “protection by exclusion” is largely driven by ambiguity aversion: when consequences are uncertain, exclusion feels safer.
COVID-19 vaccine trials followed this pattern, leaving pregnant patients and clinicians to rely on anecdotal experience and limited real-world or animal data.
A more balanced approach recognizes that:
- Evidence gaps in these populations create clinical uncertainty.
- Managed inclusion can generate data safely and ethically.
Staged approaches, like pharmacokinetic substudies and modeling, can build evidence while reducing exposure. Planning frameworks could require teams to specify what evidence will be missing at approval if pregnant or lactating women are excluded, and who will bear that risk. Framing staged inclusion pathways as risk containment aligns with current dose escalation and safety margin strategies.
Reframing the question from “Can we justify exclusion?” to “Can we justify ignorance?” changes the decision-making pathway.
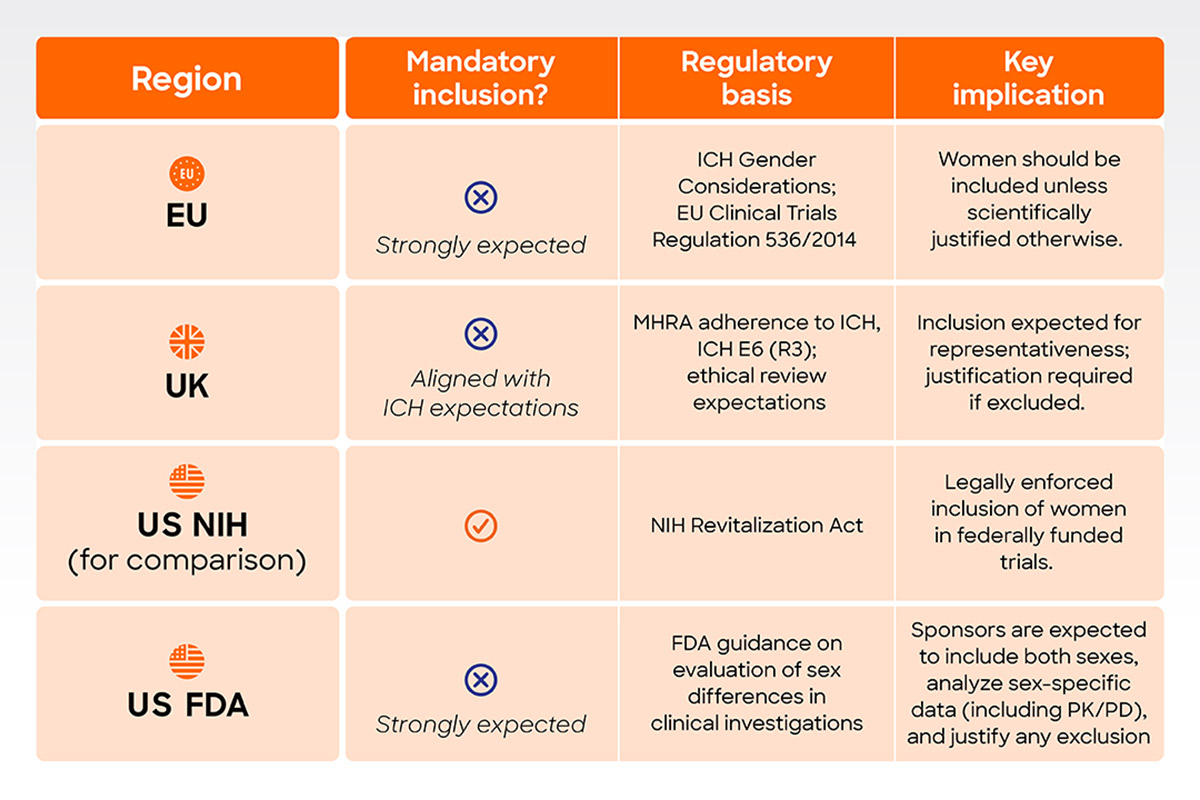 Table 1. Summary of regulatory expectations and requirements for inclusion of women in clinical trials across key jurisdictions.
Table 1. Summary of regulatory expectations and requirements for inclusion of women in clinical trials across key jurisdictions.Why Has Guidance Not Translated into Consistent Practice?
In the US, regulatory guidance has evolved to include expectations for studying sex differences, but behavior has not always followed suit. Feedback on sex-specific evidence is often delayed or diffuse, which is driven by three factors:
- Inconsistent enforcement: Guidance is not always applied uniformly, allowing variation in how sex-specific data is generated and presented.
- Incentives favor speed and predictability: Sponsors optimize for timelines, cost, and regulatory certainty.
- Diffused accountability: Responsibility for inclusion and analysis is spread across sponsors, regulators, ethics committees, and journals. Tools like regulatory pre-mortems—for example, asking, “If this product required a sex-specific label change in five years, what would we wish we had designed differently?”—could help. Instead, teams often default to acceptable and convenient approaches rather than those that generate the most complete evidence.
Unclear or inconsistent expectations make individuals rely on precedent. Limited feedback slows learning.
Behavior changes when feedback is timely, specific, and linked to future outcomes. Stronger feedback loops can help:
- Clear regulatory signals on expectations for sex-specific evidence
- Consistent review feedback highlighting gaps
- Internal metrics to track evidence quality across development programs. Highlighting positive deviations—programs that planned well and avoided downstream corrections—helps establish new norms and reinforces the value of early, sex-specific planning.
These mechanisms align incentives with desired outcomes without adding unnecessary burden. Advanced analytics, including AI, can support regulatory evaluation of safety and efficacy. But without consistent sex-specific data generation requirements, even sophisticated tools cannot address fundamental evidence gaps.
What This Means for Patients, Regulators, and Sponsors
The consequences of incomplete sex-specific evidence extend beyond medicines development. A lack of robust sex-specific evidence creates measurable consequences throughout a product’s lifecycle.
For patients, it affects safety, dosing accuracy, and confidence in treatment decisions. Unrepresentative data increases uncertainty.
For regulators, it creates downstream challenges:
- Safety signals may emerge post-approval.
- Labels may require revision.
- Inconsistencies in the availability, quality, and use of sex-specific evidence may arise across therapeutic areas.
For sponsors, the impact is clinical and commercial:
- Post-marketing changes introduce additional cost and complexity.
- Reputational risk increases when safety issues emerge.
- Development inefficiencies arise from avoidable rework.
From Expectation to Standard Practice
If regulators want sex-specific evidence, they must make it a predictable requirement, not an aspiration. What can support this shift?
- Regulators: Set clear, consistent expectations for sex-specific analysis and apply them throughout review processes.
- Sponsors: Ensure sex-specific design and analysis are embedded as core components in early development, not as add-ons.
- Research journals and funders: Reinforce reporting standards and incentivize high-quality, sex-disaggregated evidence.
EU Regulation: Regulation No 536/2014 of the European Parliament on clinical trials on medicinal products for human use
UK Regulation: Medicines for Human Use (Clinical Trials) Regulation 2025
US: NIH Revitalization Act
US: FDA Clinical Trials Guidance Documents
The next article will examine how decisions are made and how these barriers can be reshaped to close the gap.