

Richmond Pharmacology
Alpharmaxim Healthcare Communications
Behavior Change Consultant
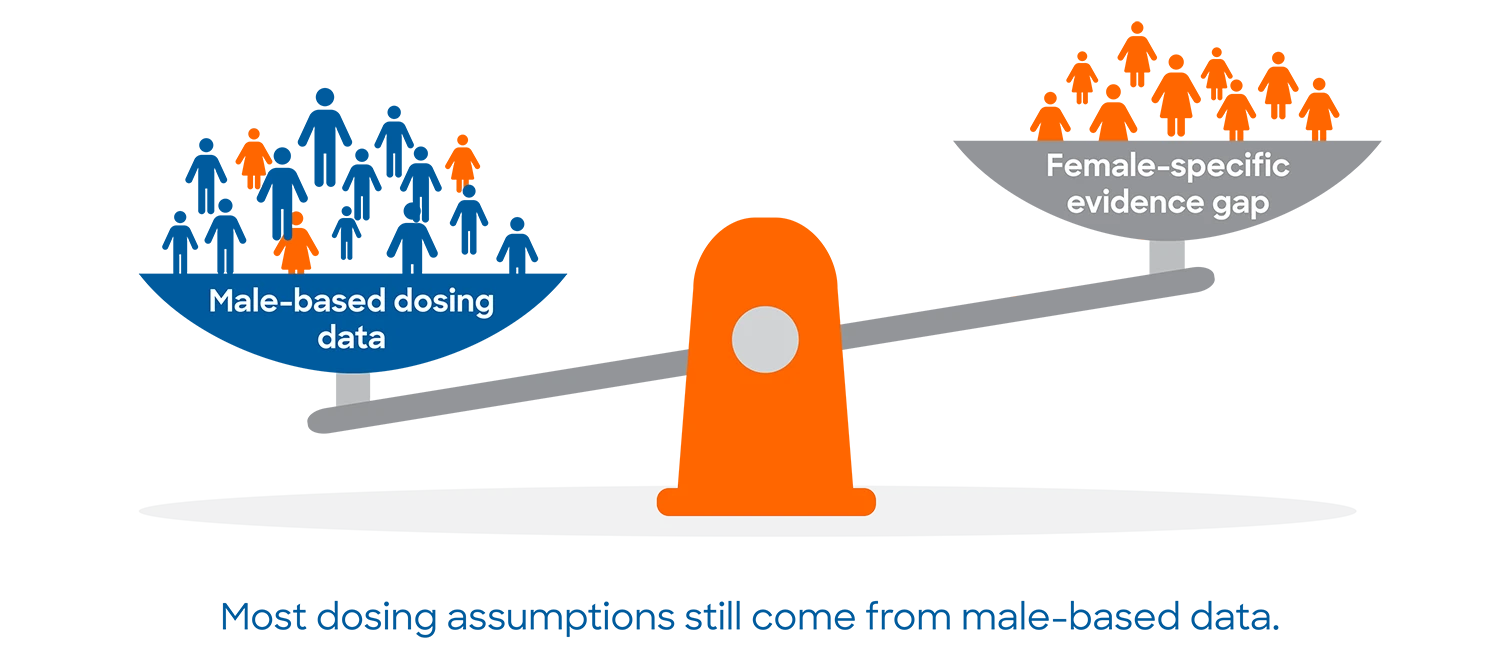
espite decades of guidance encouraging the inclusion of women in clinical research, drug development continues to rely on evidence that is disproportionately generated from male participants. The result is a persistent female data deficit that affects safety, dosing, efficacy, and trust in medicines.
Understanding and changing these behaviors is essential if the industry is to move from intention to impact.

How Historical Decisions Shaped Today’s Data Practices
The reactionary regulatory recommendation to exclude all women of childbearing potential from early-phase clinical trials following the thalidomide tragedy is frequently cited as the origin of the problem. While the intention was to protect women and unborn children from harm, the behavioral legacy was far broader. Over time, omission remained the default rather than a risk-managed exception, despite regulatory guidance.
Instead of asking how women could be safely included, development programs learned to ask whether inclusion could be avoided. This pattern was reinforced through regulatory caution and operational norms. What began as a response to a specific risk hardened into routine practice.
From a behavioral perspective, this pattern reflects well-described heuristics that shape decision making in often complex environments. Default bias (the human tendency to stick with a pre-set option, even when an alternative might be better) can encourage teams to repeat established trial designs; loss aversion (where the fear of possible negative consequences dominates rational evaluation of potential gains) magnifies perceived risk over potential value; and status quo bias (the tendency to prefer things to stay the same, even when change could lead to better outcomes) reinforces the comfort of historically “acceptable” populations.
Today, most sponsors recognize the importance of gender-specific data. Yet many programs still default to male-dominated populations, particularly in early development, because that is what teams are used to doing. A comprehensive review of 86 FDA-approved drugs showed that most dosing regimens were derived from male-based pharmacokinetics. Women experienced adverse effects in over 90% of cases due to slower elimination and higher exposure.
Why Awareness Has Not Changed Data-Collection Behavior
There is no shortage of awareness about the female data gap. The problem is that awareness alone does not change behavior.
Sustainable change depends on whether people have the capability to act differently, the opportunity to do so within existing systems, and the motivation to challenge established norms. In drug development, these three elements interact to sustain male-default trial designs, even among teams that intellectually support inclusion. These three forces are important when considering data-collection decisions in clinical trials.
- Capability. Many teams lack confidence in how to design studies that meaningfully collect and analyze gender-specific data.
- Opportunity. Trial infrastructure is often not designed around women’s lives. Rigid visit schedules, lack of childcare support, limited flexibility for caregivers, and site selection that does not reflect community demographics all reduce the opportunities for female participation.
- Motivation. If teams believe that regulators will accept male-default data, or that gender-specific analysis is unlikely to change approval outcomes, there is little motivation to challenge existing practice.
These forces combine to produce a system in which the female data deficit is not the result of active exclusion but of passive default.
What This Means for Data Collection in Practice
If the problem is behavioral, then the solution lies in changing how decisions about data collection are made and supported. How? By diagnosing the behavior before designing the trial.
Whether female representation is improved in practice depends on which unique behavioral barriers are present at each decision point. Some teams struggle with psychological capability (uncertainty about gender-specific design), others with opportunity (trial environments that unintentionally exclude women), and others with motivation (perceived misalignment between inclusion and success metrics). Without diagnosing which of these barriers exist, well-intentioned guidance can risk being ignored.
First, gender should be treated as a core design variable at the point of protocol development and not at secondary or post hoc analysis. This means asking early, at the protocol design stage, which decisions will be informed by gender-specific data. If dosing, safety, or efficacy may differ, then trials must be designed to generate that evidence, not retrofitted after the fact. This establishes a default expectation of gender-aware trial design, reducing reliance on retrospective rationalization or incomplete subgroup analysis.
Second, early-phase studies matter. Many of the most consequential data gaps originate in phase 1, where dosing and exposure assumptions are set. Designing early studies with gender-stratified cohorts, even at modest scale, can prevent downstream safety issues and reduce reliance on post-marketing correction.
Third, inclusion should be operationally enabled, not just encouraged. Practical adjustments such as flexible visit windows, decentralized assessments, reimbursement for childcare or travel, and engagement with patient advocacy groups can materially improve recruitment and retention of women. These environmental restructuring interventions remove frictions that disproportionately affect women’s participation. By changing the context in which participation occurs, inclusion becomes easier without requiring additional motivation or persuasion. These are not abstract commitments; they are concrete design choices that shape who generates the data.
Fourth, pregnancy risk should be managed, not avoided. Blanket exclusion of women of childbearing potential or pregnant women removes the possibility of learning how medicines behave in populations that will ultimately use them. Staged inclusion, clear governance, and transparent consent processes allow data to be generated ethically rather than deferred indefinitely.
The Regulatory Role in Reshaping Behavior
Regulation plays a critical role not only in setting expectations but also in shaping behavior. Clear, consistent regulatory signals reduce uncertainty and change what teams perceive as acceptable and worthwhile. When regulators expect gender-disaggregated data, challenge unjustified exclusions, and value evidence that reflects real-world populations, sponsor behavior follows.
Equally important is enforcement. Guidance without accountability allows habitual practice to persist; thus, where deviations from guidance occur, clear justifications (based on science) should be provided. Data collection improves when expectations are not only articulated but also embedded into review, feedback, and approval processes.
From Deficit to Design Choice
The female data deficit is often discussed as a historical problem. In reality, it is a present-day drug development design choice, reinforced by behavioral defaults.
Closing the gap does not require reinventing drug development. It requires redesigning how decisions about data collection are made, supported, and rewarded. Validated behavioral frameworks are already used to address prescribing behavior where gender can be one of many barriers hindering adoption. Applying the same rigor upstream to trial design represents a natural next step. When teams are equipped with the capability, opportunity, and motivation to include women meaningfully, improved frameworks and interventions should follow.
The question is no longer whether women should be represented in drug development. It is whether the drug development landscape is designed to make that representation the norm rather than the exception.